El futuro es hoy (Cuando el destino nos alcanzó)
Enrique Alejandro Reynaud Garza
El jueves 16 de noviembre del 2023, fue uno de los días más trascendentes en la historia de la humanidad; ese día se aprobó en el Reino Unido la primera terapia génica para humanos basada en el sistema CRISPR-Cas9, unas semanas después la Administración de Drogas y Comida de estados Unidos (FDA por sus siglas en inglés) también la aprobó [1,2] (Figura 1), inaugurando de manera inequívoca e irreversible la nueva era de la ingeniería genética [3]. Resulta interesante que este hito se haya logrado, exactamente (en el mes de noviembre) cuando se cumplen 50 años de que se publicó el primer artículo que reportó la posibilidad de recombinar ADN en bacterias [4].
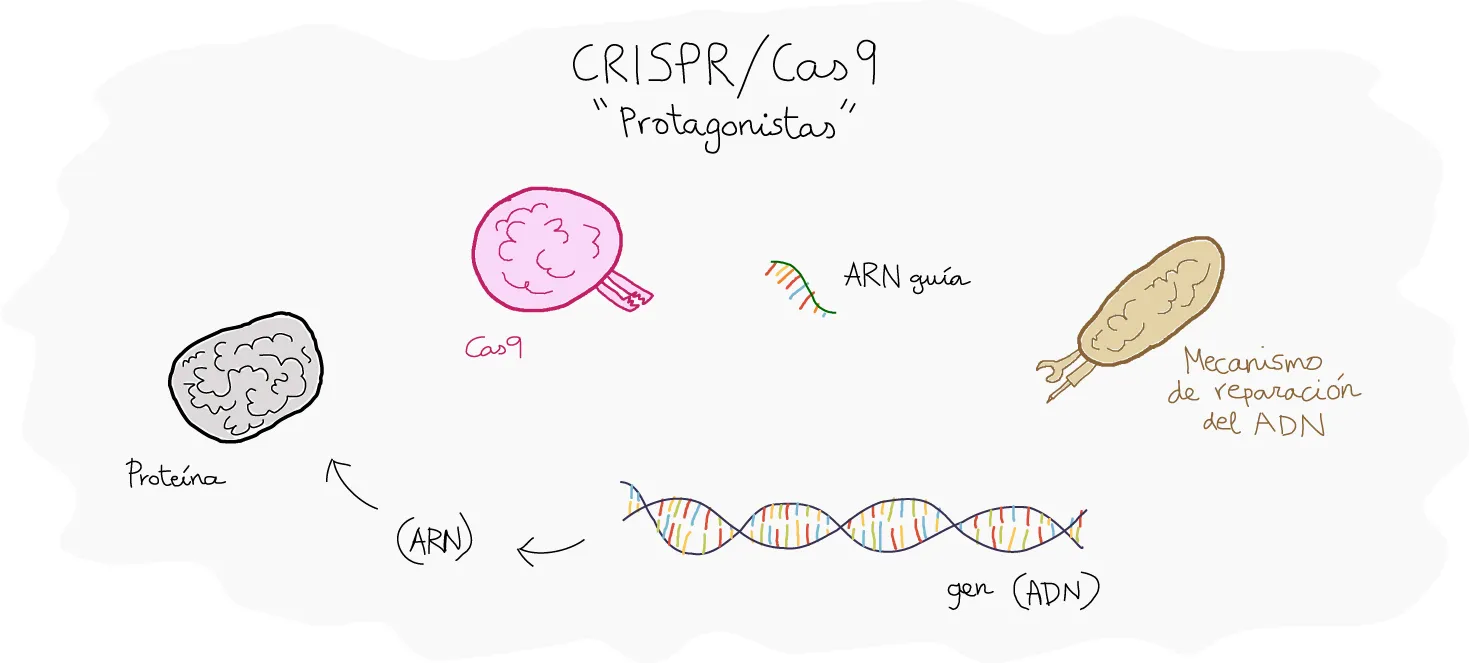
Figura 1. Presentación de los protagonistas: CRISPR/Cas9. El ARN guía es el pedacito de ARN que los investigadores utilizan para buscar el gen del ADN donde quiere causar una mutación, como el patrón. Cas 9 es una tijera enzimática, que actúa cortando el genoma donde el ARN guía encuentra a su ADN homólogo (que se parece mucho a él). Ilustración: Juan Francisco Abenza (Integrative cell and tissue dynamics).
¿Qué significa todo esto? ¿Por qué es tan trascendente? ¿Qué implica para nuestro bienestar? ¿Cómo va a ser el futuro de la humanidad? Pongámonos en contexto:
¿Qué es la terapia génica?
El genoma es la secuencia de ADN (o de ARN en el caso de algunos virus) que tiene las instrucciones para construir a un organismo. En el caso de los humanos, el genoma mide 3,000,000,000 (tres mil millones de pares de bases, que son los constituyentes del ADN). Cada célula de nuestro cuerpo tiene dos copias del genoma humano. Una copia de ellos la heredamos vía materna (a través del óvulo) y la otra vía paterna (a través del espermatozoide). Cuando se comparó el genoma de todos los humanos entre sí, se descubrió que somos prácticamente idénticos los unos con los otros, habiendo una diferencia de menos del 0.5% al comparar a cualquier par de individuos humanos; interesantemente, la copia materna y la copia paterna de nuestro genoma también difieren en ese 0.5%.
Gracias al fenómeno de la recombinación gamética, ningún genoma es idéntico a otro. Sólo con la excepción de los genomas de los gemelos idénticos, que compartieron los mismos gametos (cuando un embrión creado por un único óvulo y un único espermatozoide se separa en dos, se generan gemelos genéticamente idénticos y, técnicamente, son una clona el uno del otro). Esa diferencia azarosa del 0.5% entre un genoma y otro, es lo que nos hace individuos.
Dentro de la secuencia del genoma de un organismo existen regiones que codifican ARNs funcionales, cuya función no vamos a discutir en este espacio. También hay aproximadamente 20 mil genes que codifican proteínas distintas. La mayoría de las funciones celulares son llevadas a cabo por proteínas, así, si un gen que codifica para una proteína tiene una mutación, este puede codificar para una proteína defectuosa, la cual no puede hacer su trabajo o incluso se puede volver tóxica, causando una enfermedad genética.
La mayoría de las diferencias entre los pares de bases de nuestros genomas (polimorfismos de un solo nucleótido o SNPs, por sus siglas en inglés), no tienen ningún efecto en nuestra salud o desarrollo; es decir, son neutras. Sin embargo, una minoría de ellas puede afectar la secuencia codificadora de un gen, haciéndolo codificar para una proteína defectuosa y por lo tanto puede causar una enfermedad genética.
No los quiero espantar, pero, en promedio, cada uno de nosotros es portador de al menos 5 mutaciones recesivas letales y otras muchas no letales, pero que pueden tener consecuencias médicas. Una mutación recesiva sólo tiene efecto cuando tanto la copia materna como la copia paterna, son mutantes.
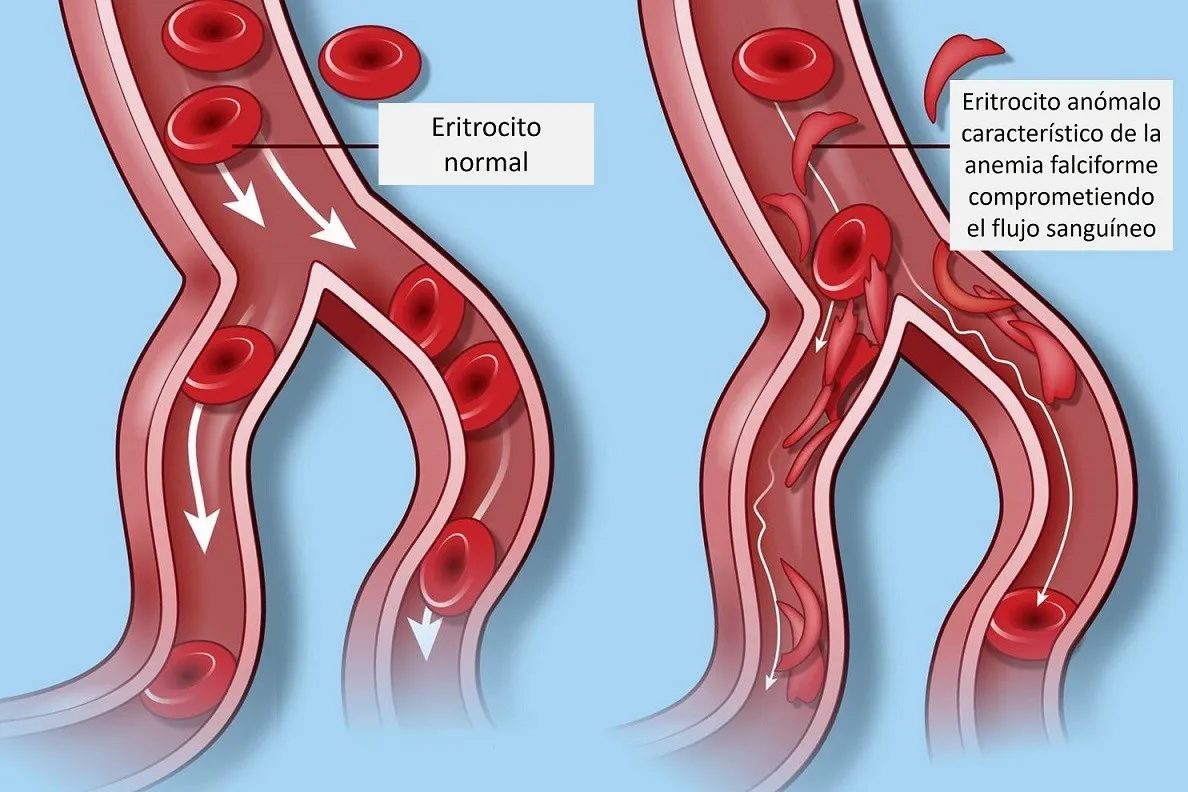
En la mayoría de los casos, ser portador de una mutación recesiva no tiene consecuencia porque al tener dos copias de nuestro genoma -con pequeñas diferencias entre sí-, estas mutaciones casi nunca son las mismas, por lo que andamos por la vida sin darnos cuenta de ellas. El problema empieza cuando desafortunadamente heredamos la misma mutación en el mismo gen, tanto en nuestro genoma materno como en el paterno; es decir, mientras seamos heterocigotos (una copia normal y una mutante de un mismo gen), no hay problema. Sin embargo, al ser homocigoto (dos copias mutantes), se manifiesta la enfermedad genética. Este es el caso de la mayoría de las enfermedades de los síndromes genéticos monogénicos (atribuibles a un gen único) como son la fibrosis quística, la fenilcetonuria, la enfermedad de Tay-Sachs, el mal de Huntington, la anemia falciforme (Figura 2) y la beta-talasemia.
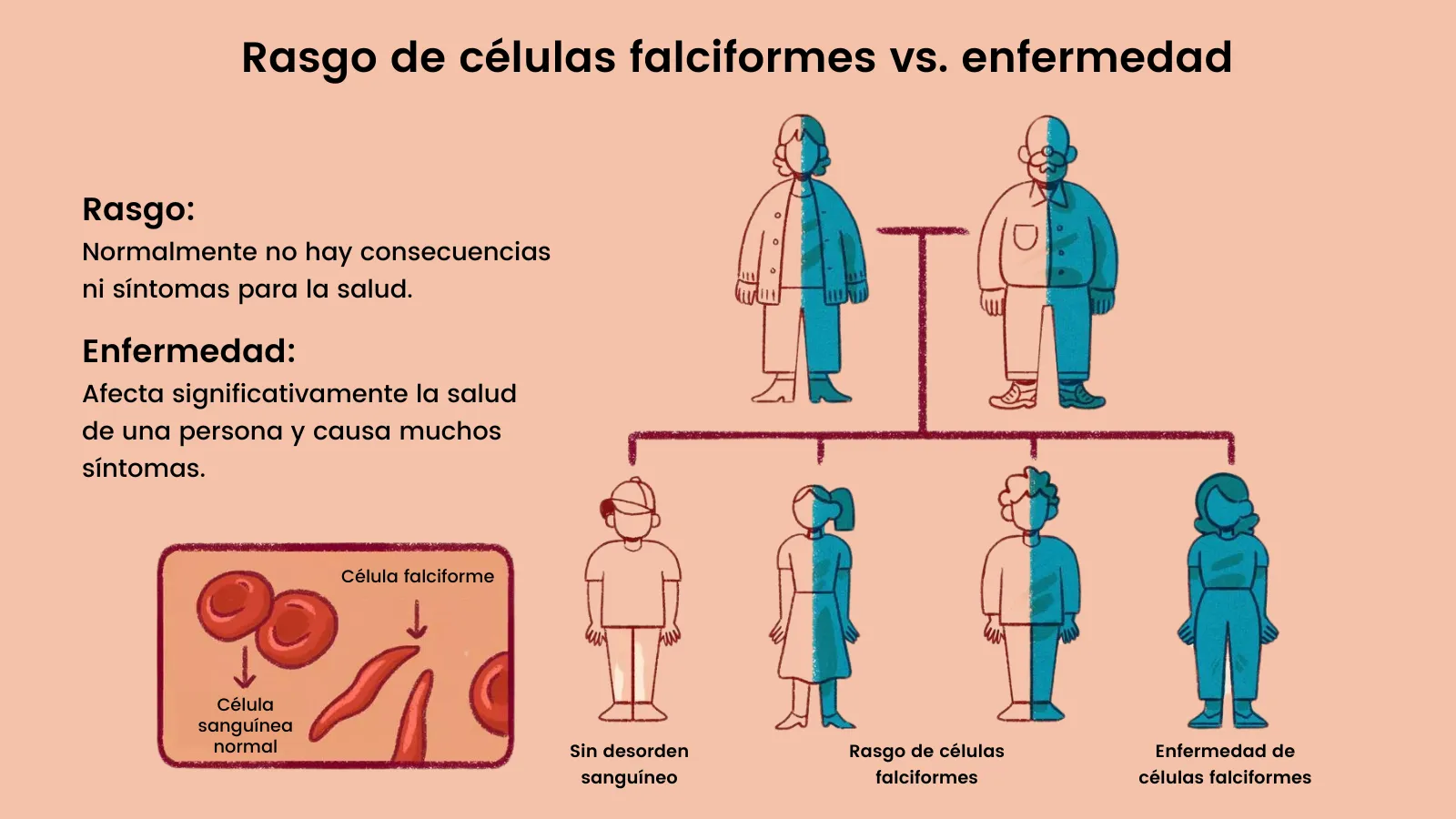
Figura 2. Manifestación de la enfermedad genética en la anemia falciforme. Ilustración modificada de Theresa Chiechi (VerywellHealth).
La terapia génica significa sustituir un gen mutante por su versión normal, “rescatando” de esta manera la función génica, y de esta manera evitar los efectos patológicos de la mutación. Esto se ha intentado y en algunos casos se ha logrado [5]. Sin embargo, siempre hay limitaciones técnicas muy complejas. La principal y más grande es cómo introducir la versión normal del gen. La segunda es cómo hacerlo en el tejido correcto y en el lugar correcto del genoma.
La primera es una limitación grande y compleja, pero se ha podido resolver utilizando técnicas quirúrgicas muy ingeniosas combinadas con métodos cada vez más eficientes de transmisión génica, en las que se utilizan métodos químicos o virus manipulados genéticamente, para transferir construcciones génicas (esto es, fragmentos de ADN recombinante) dentro de nuestro organismo. En pocas palabras: fuerza bruta. Por otro lado, hay que tener en cuenta que, si introducimos ADN a nuestras células, los mecanismos de protección del genoma (que todas nuestras células tienen), pueden contender con él, insertándolo de manera aleatoria dentro de nuestro genoma. La inserción de secuencias al azar son mutaciones por sí mismas y si se insertan dentro de otro gen, pueden causar nuevas enfermedades, tales como cáncer.
Por si esto fuera poco, el contexto del genoma (contexto cromosomal) también puede afectar. Si nuestro gen terapéutico se expresa (esto es, como se prende o se apaga) bien o mal, o en el tiempo incorrecto, esto disminuye o nulifica la eficiencia de la terapia génica. Por ejemplo, la hemoglobina sólo puede y debe expresarse en los eritrocitos y en ningún otro tipo celular.
Si lo piensan, este es un problema mayúsculo, ya que el gen blanco que se quiere sustituir en el genoma es una fracción infinitesimal del genoma. Recordemos que la secuencia codificadora de un gen promedio es de aproximadamente mil nucleótidos y el genoma tiene tres mil millones de pares de bases de nucleótidos. O sea, que se necesita una tecnología capaz de atinarle a un blanco minúsculo, si queremos sustituir un solo nucleótido mutante o SNP, el blanco es de uno en tres mil millones. Hace diez años una tecnología así era ciencia ficción.
CRISPR-Cas9
En el año 2012, Jennifer Doudna de la Universidad de California, Berkeley y Emmanuelle Charpentier quien es ahora investigador del Instituto Max Plank de Biología de las Infecciones en Berlín, publicaron un artículo en donde demostraban que la enzima bacteriana Cas9 podía ser programada cambiando la secuencia de ARN asociada a ella, para que cortara sitios específicos en el ADN, estableciendo las bases teóricas y moleculares del sistema conocido como CRISPR–Cas9 [3].
Cas9 es una proteína bacteriana cuya función molecular es cortar el ADN en sitios específicos, determinados por una secuencia de ARN llamada “guía”. Importantemente, Cas9 es programable y el sitio de corte lo podemos definir arbitrariamente cambiando la secuencia guía. De esta manera podemos hacer cortes en nucleótidos específicos en el genoma de un organismo tan complejo como el ser humano. En el año 2020, Jennifer Doudna y Emmanuelle Charpentier (Figura 3) fueron galardonadas con el premio Nobel de química por haber descubierto el sistema CRiSPR-Cas9 que permitía hacer edición precisa del genoma.

Figura 3. Emmanuelle Charpentier y Jennifer Doudna galardonadas con el premio Nobel de química en 2020, por el desarrollo de un método para edición del genoma. Ilustración: Niklas Elmehed.
¿Cómo se edita el genoma utilizando Cas9?
De manera muy simplificada, lo que se hace es que se programa a Cas9, usando una guía que la dirige al nucleótido que se quiere modificar. Esto causa un corte único en la secuencia de doble cadena del ADN.
Las células cuidan muchísimo la integridad de su genoma y uno de los peores insultos es un corte a la doble cadena, ya que desintegra a los cromosomas. Por ello, la maquinaria de reparación del genoma responde de manera inmediata para reparar -cueste lo que cueste- esta doble ruptura. El problema es que la maquinaria no sabe si se perdió o no, un segmento del genoma en la ruptura, y lo arregla "como puede", esto es, al azar o de forma no controlada, ya que es una emergencia, esto debido al mecanismo de reparación, que casi siempre lleva a una pérdida de nucleótidos alrededor del sitio de ruptura, con la consiguiente pérdida de la información genética. Esto puede ser útil si se quiere anular la función de un gen en particular.
Alternativamente, la maquinaria de reparación puede copiar e insertar nuevas secuencias de ADN en el sitio de ruptura, si se le provee una secuencia que pueda copiar. Entonces, cuando se induce el corte y al mismo tiempo se provee la secuencia del gen normal, la maquinaria “lee” la secuencia normal del gene y la inserta en el sitio que se cortó, sustituyendo la secuencia mutante por la normal (también llamada “silvestre”) (Figura 4). Con estas dos estrategias es posible apagar genes, o reintroducir dentro de la célula la función de un gen defectuoso.
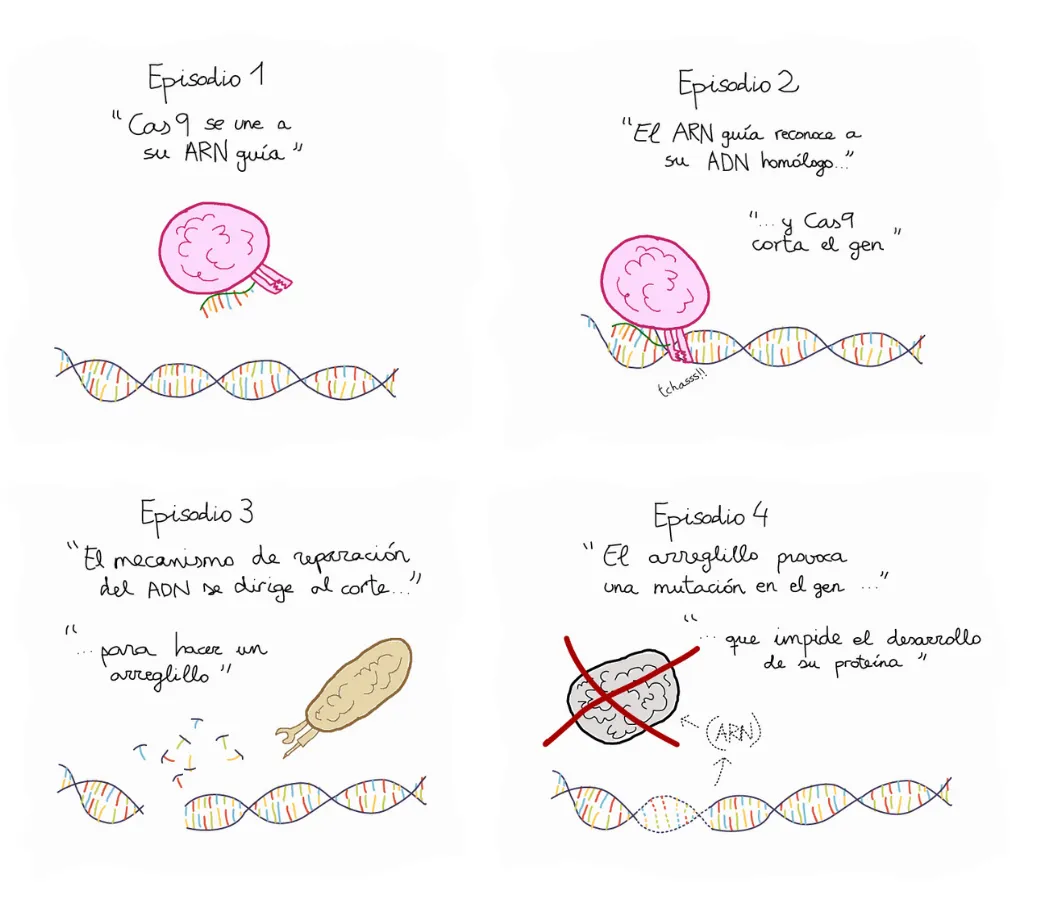
Figura 4. Funcionamiento de CRISPR-Cas9 en 4 episodios. Ilustración: Juan Francisco Abenza (Integrative cell and tissue dynamics).
¿Cuál fue la terapia génica que se aprobó en el Reino Unido?
Cuando respiramos, el oxígeno del aire es transportado a nuestros órganos internos por las células rojas de la sangre (eritrocitos). La proteína transportadora del oxígeno es la hemoglobina. En los adultos esta proteína está conformada por cuatro subunidades proteicas llamadas globinas: dos alpha (α) y dos beta (β) (Figura 5). Hay un gen que codifica para las subunidades α-globina y otro gen que codifica para las subunidades β-globina.
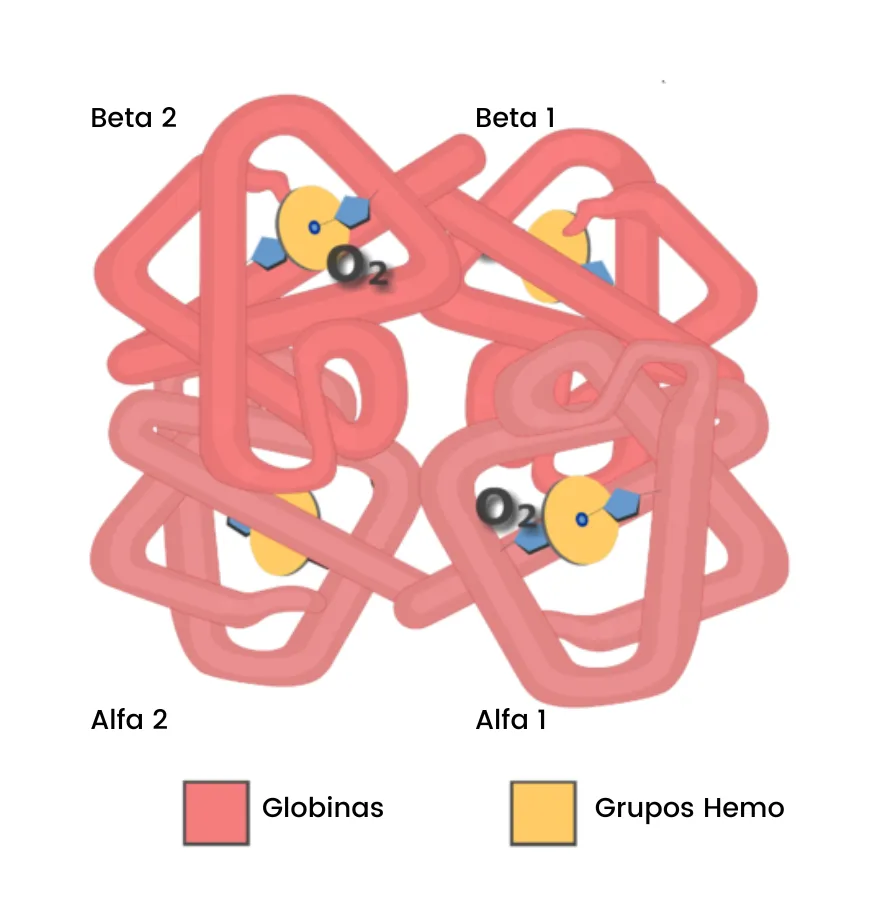
Figura 5. Estructura de la hemoglobina mostrando sus cuatro subunidades proteicas llamadas globinas: dos alfa (α) y dos beta (β).
Durante el desarrollo fetal, los humanos expresamos (esto es, que un gen se prenda), en lugar de la globina β adulta, una forma especial de esta, llamada HbF, que sustituye de manera funcional a la forma β. La hemoglobina que tiene HbF es más afín por el oxígeno que la forma adulta, ya que, mientras somos fetos, tenemos que “robarle” el oxígeno a la sangre de nuestras madres, a través de la placenta. Cuando nacemos, una proteína codificada por el gen BCL11A, se encarga de apagar la expresión (producción) de la hemoglobina HbF y se induce la expresión de la globina β.
Existen dos enfermedades asociadas a defectos en el gen de la globina β, la anemia falciforme y la β-talasemia. La anemia falciforme es una enfermedad recesiva que en Estados unidos afecta a 4 de cada mil personas de ascendencia africana. Esta enfermedad es causada por una mutación en el gen de la β-globina (proteína que forma parte de la hemoglobina), cambiando su estructura de manera irreversible a una forma no funcional cuando no está asociada al oxígeno, haciendo que los eritrocitos no transporten el oxígeno de manera eficiente y cambiando la forma de estos de redondos a forma de hoz. Estos eritrocitos defectuosos se atoran en los vasos capilares, en el bazo, el hígado y otros órganos, causando crisis de dolor acompañado por daño tisular. Si no es tratada, puede ser mortal y, aún tratada, reduce la calidad y la esperanza de vida de los pacientes que la sufren.
La β-talasemia es una enfermedad genética recesiva que afecta a 1 de cada 100 mil individuos y se caracteriza por la deficiencia o ausencia de la producción de la β-globina. Es una anemia genética y también reduce de manera dramática la calidad y la expectativa de vida de los pacientes.
Tanto la anemia falciforme como la β-talasemia son enfermedades -hasta ahora incurables-, que requieren que los pacientes reciban transfusiones sanguíneas periódicas, así como otras intervenciones farmacéuticas y médicas. El tratamiento es extremadamente costoso y los síntomas son muy dolorosos e incapacitantes.
La terapia génica aprobada en el Reino Unido el 16 de noviembre de 2023 es capaz de curar, tanto a la anemia falciforme, como a la β-talasemia y consiste en la siguiente estrategia: Primero se extraen células de la médula ósea de los pacientes. En esta se encuentran las células madre que van a dar origen al linaje eritroide, las células del linaje eritroide después de un proceso de diferenciación y maduración se convierten en eritrocitos (las células madre son células precursoras que tenemos en nuestro organismo y su función es reponer a las células funcionales conforme estas van envejeciendo y muriendo; tenemos células madre para todos nuestros tejidos). Después, a estas células se les introduce Cas9, programada con un ARN guía que la dirige al gen BCL11A, destruyéndolo, de manera que ya no puede reprimir al gen de la hemoglobina HbF, la cual sustituye la función de la β-globina. Esto provoca que los eritrocitos de los pacientes mantengan su forma y función. Una vez que los precursores eritroides han sido modificados, se re-transplantan al paciente (lo que se llama un trasplante autólogo), lo que evita cualquier riesgo de rechazo.
En las pruebas clínicas, prácticamente todos los pacientes se curaron y sus síntomas desaparecieron, al parecer de manera permanente. Es muy probable que esta terapia génica se apruebe en Estados Unidos en los próximos meses.
¿Por qué es tan importante que se haya aprobado una terapia génica basada en el sistema CRISPR-Cas9?
El sistema CRISPR-Cas9 es un sistema universal de reprogramación y modificación de genomas. Esto lo hace equivalente a la invención de los sistemas operativos y los lenguajes de programación de alto nivel de las computadoras, que permiten que prácticamente cualquier persona se convierte en un programador y usuario de las computadoras. Como sabemos, pocas tecnologías han tenido un impacto tan grande y universal en el crecimiento económico y en la estructura social como fue la invención de las computadoras personales, el internet, los teléfonos móviles y las redes sociales.
CRISPR-Cas9 va a traer una revolución similar a la de las computadoras, pero en el mundo biológico. Para empezar, esta tecnología o alguna otra similar van –eventualmente- a erradicar todas las enfermedades de origen monogénico y probablemente a muchas de origen multigénico (que son más difíciles de atacar, porque hay que hacer modificaciones en varios genes y la contribución de cada uno de estos a la enfermedad no es tan obvia).
Por otro lado, abre la posibilidad de modificar la línea germinal humana de manera permanente para adquirir o perder características, acelerando la evolución biológica de nuestra especie (aún no tenemos claro cuáles pueden ser las consecuencias de esto). Además, no solo se están modificando humanos, sino que también otros organismos. Por ejemplo, hay compañías que están modificando puercos con modificaciones en al menos 62 lugares independientes de su genoma y ya se han hecho trasplantes experimentales de riñones de estos puercos a personas con muerte cerebral, para demostrar que no hay rechazo y que los riñones son funcionales. Asimismo, ya se han hecho dos trasplantes de corazón de cerdo a personas con deficiencia cardiaca. El primer paciente vivió dos meses, y el segundo seis semanas, con corazones de cerdo. Parece poco, pero hay que tomar en cuenta que es una tecnología en pañales.
Hay compañías que están desarrollando complejos especializados para la producción masiva de estos órganos y calculan que van a poder producir miles órganos para trasplantes humanos al año. Esto significa que, en el plazo de una o dos décadas, ya no habrá escasez de órganos para trasplantes, lo que podrá alargar la vida de mucha gente. Finalmente, el impacto biotecnológico de esta tecnología va a ser incalculable, ya que va, desde tener cultivos y ganado mejorado que sea resistente a enfermedades y al cambio climático, a mejorar la productividad agrícola y ganadera, mejorando la calidad de los alimentos y disminuyendo su costo e impacto económico.
A largo plazo, la ingeniería genética mediada por estas tecnologías tiene el potencial de revivir especies extintas como el mamut y el pájaro dodo, así como plantas, invertebrados y microorganismos que nos permitirán renovar y mejorar a los distintos ecosistemas del planeta. Todo esto mejorará la ecósfera planetaria y la calidad de vida nuestra especie.
Lo interesante apenas comienza.
REFERENCIAS
- Wong C. (16 de noviembre de 2023). UK first to approve CRISPR treatment for diseases: what you need to know. Nature.
- Corless V. (21 de noviembre de 2023). CRISPR gene therapy for sickle cell disease and β-thalassemia gets UK approval. Advanced Science News. https://www.advancedsciencenews.com/crispr-gene-therapy-for-sickle-cell-disease-and-β-thalassemia-gets-uk-approval/
- Reynaud E. (2016). Bienvenidos a la nueva era de la Ingeniería Genética. Biotecnología en Movimiento; 5:23-27.
- Cohen, S. N., Chang, A.C.Y., Boyer, H. W., Helling, R. B. (1973) Construction of biologically functional bacterial plasmids in vitro, Proceedings of the National Academy of Sciences USA, Vol. 70, No. 11, pags. 3240-3244, November.
- Reynaud E. (2018). El niño de la piel transgénica. Biotecnología en Movimiento; 13:27-29.
Comparte este artículo en redes sociales

Acerca de los autores
Enrique Reynaud es investigador del Instituto de Biotecnología de la UNAM. Cursó toda su educación superior en la UNAM; es Lic. en Investigación Biomédica Básica, Maestro en Biotecnología y Dr. en Investigación Biomédica Básica. Posteriormente hizo un postdoctorado en Genética del comportamiento en la universidad de Stanford con el Dr. Bruce Baker con una beca de “ Fellow in Biomedical Sciences” de la “ PEW Charitable trust”. Ha formado cuatro doctores, doce maestros en ciencias y ha dirigido 12 tesis de licenciatura. Fungió como presidente del comité de Bioética del Instituto de Biotecnología por cinco años. Actualmente hace investigaciones de Neurobiología del Desarrollo en la mosca de la fruta Drosophila melanogaster utilizándola para estudiar el desarrollo del sistema nervioso y enfermedades neurodegenerativas tales como el mal de Parkinson.
Contacto: enrique.reynaud@ibt.unam.mx